
The Longevity & Aging Series is a collaboration brought to you by Aging (Aging-US) and FOXO Technologies. This monthly series of video interviews invites Aging researchers to speak with researcher and host Dr. Brian Chen. Dr. Chen is an adjunct faculty member at the University of California San Diego and the Chief Science Officer at FOXO Technologies.
—
Below is a transcription of the fourth installment of the Longevity & Aging Series, where Drs. Carly Bobak, Cristian Coarfa, and Andrew DiNardo detail their research paper published by Aging (Aging-US), entitled, “Increased DNA methylation, cellular senescence and premature epigenetic aging in guinea pigs and humans with tuberculosis.”
All right. Thank you everybody for joining us. I’m Brian Chen, Chief Science Officer at FOXO Technologies, here on behalf of Aging. Today, we have a very exciting paper titled, “Increased DNA methylation, cellular senescence and premature epigenetic aging in guinea pigs and humans with tuberculosis.” Here today we have three of the authors, and I’m going to have them go through, in order, to introduce themselves. So Andrew, do you want to start?
Sure. I am Andrew DiNardo, I am an Assistant Professor in Infectious Disease here at Baylor College of Medicine.
Great, and then Cristian.
Hey, I’m Cristian Coarfa, I’m an Associate Professor at the Baylor College of Medicine in the Cancer Center and my main focus is multi-omics analysis.
All right, and Carly.
Hi. So my name is Carly Bobak, I’m a lecturer and researcher in Biomedical Data Science at Dartmouth College, and a lot of this work was part of my PhD thesis, also done at Dartmouth College.
Great. Can one of you guys give us a background about what this paper is about, set the stage for us?
Sure. We previously had done work in DNA methylation in schistosomiasis in adolescence from Eswatini with schistosomiasis and then adults from Eswatini, formerly Swaziland. We previously studied the effect of DNA hypermethylation on the immune system. Carly and I had the pleasure of meeting at a conference and we found out that we had a lot of overlap between our research interests. From there, we started a collaboration that led to this paper.
Perfect. And Andrew, maybe you’re in a good position to say this, but cellular senescence, premature aging, DNA methylation. These are all aging-specific things, and then you study infectious diseases. So can you talk about the marriage between the two different fields?
Well, there’s been this hypothesis that severe or chronic infections induce premature aging, and there’s a little bit of data on that for CMV, there’s a little bit more data for HIV, but there’s really no data for any other infections, including tuberculosis, which is the most common cause of pneumonia. Before the pandemic, the most common cause of infectious disease mortality was due to tuberculosis. We don’t have data on sepsis, on pneumonia. There’s a lot of infections that we are still lacking data, despite this hypothesis and this assumption that infections lead to cellular senescence and premature cellular aging.
Perfect. Great. Now that we’ve set the stage, why don’t you guys take it away and show us what you learned.
Well, our previous paper that I briefly mentioned here in tuberculosis identified that TB patients have DNA hypermethylation in the PI3K pathway, the TNF, the IL-2-STAT5, the interferon-gamma signaling pathways. And that these DNA hypermethylation marks were associated with decreased immune responsiveness, both to BCG stimulation, both to TB stimulation, also to interferon-gamma stimulation, also IL-12 and mitogen. So it was an across the board, both antigen-specific and antigen-nonspecific immune hyporesponsiveness.
So in this study we wanted to know if these individuals… One of the first things we wanted to ask is: Were these healthy individuals that had detrimental epigenetic marks, these DNA hypermethylation marks, and that predisposed them to develop TB – which is certainly possible – or if TB in and of itself did that? We were able to collaborate with Jeff Cirillo at Texas A&M to ask that question using a guinea pig model. And then the latter part is: What are the consequences of these epigenetic changes, both for sepsis, for COVID, for pneumonia, and for TB after successful therapy or any of those infections? There’s a two- to three-fold increased risk of death after survival. One of the hypotheses we had is that the increased cellular senescence and the increased premature epigenetic aging, which we’ll discuss, might be leading to some of these secondary problems.
Technically, it’s nice to be part of this team because we all bring different skills sets to the table. Andrew is obviously a very passionate physician scientist, and he conducts a successful research lab. He also travels around the world, seeing patients from different countries, collecting samples, imparting knowledge and learning. And I’m very fortunate to be working with him. And then Carly, obviously, is a rising star and, actually, she brings the best of both worlds because she has a formal training both biology and bioinformatics. So she was able to bring the best of both fields. I’m focused on multi-omics analysis and working with Andrew was a pleasure because I kind of felt that, on one hand, I was able to find some interesting problems to work. On the other hand, he had enough knowledge and patience with us to actually blend those skills.
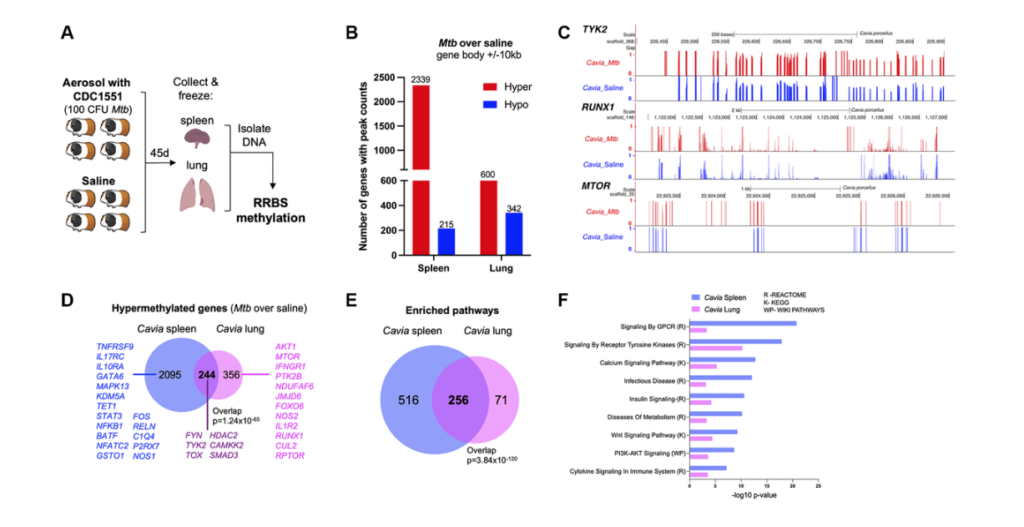
In this example, we focused on DNA methylation. Past work of our groups focused on immuno microarrays. On the other hand, this time we wanted to move into the realm of sequencing. Were able to get slightly deeper and more detailed information. So the experiment performed started with the guinea pig. They were infected with tuberculosis bacteria or with the stallion solution and after 45 days, spleen and lungs were removed. And then they were set with the process called the RRBS methylation. And RRBS stands for reduced representation bisulfite sequencing. It’s named this way to distinguish from the whole genome bisulfite sequencing. There is a significant saving in cost, which to us means that we can do more samples with it. And then technically the focus of the reducer presentation is done by using multiple digestion enzymes. So technically once we got this data, we process it to standard pipelines.
We use Bismark tool for the mapping, we use a tool called DSS to the differential methylation, and then once we get these results, we took a step back and said, “Okay, what are the questions that we try to address with this particular essay?” So one of the question was: Is this guinea pig model relevant to human disease? Okay. Another question was: Well, are the longest being both informative? And then the last one for this essay was: How does it link to previous work in DNA methylation done in human blood in particular cell types that Andrew and team have already parted on? Okay. So then we tried to address these questions one by one. So first of all, the interesting observation was that hyper methylation seemed to be a prominent female type in both spleen and lung. And then technically, as you can see from the graph on the right about 80% of the differential methylation genes in the spleen, and about 60% of those in the lung had hyper methylation.
And this is tracking with previous reports of others and Andrew showing that hyper methylation is a major epigenetics carrying phenotype into tuberculosis. Next step we wanted to do integrate with our previous data. So one question was: Are lung and spleen informative? So the answer is yes, they both indicate hyper methylation phenotype. And that also when we take genes associated with differentiated regions and put them through standard approaches of looking for pathway enrichment. We can see that genes from both tissues and reach for some of the same major pathways. So technically we have reached for cyto signaling calcium signaling metabolism, pediatric kinase, and tyrosine kinase pathways. And technically these are been reported. Each of these groups of pathways have been reported and extensively published in infectious diseases. So now one of the most important questions is excellent. We have the animal model, we have data from two tissues that seem to be an agreement with each other, they seem to be enriching for broad terms reported, but can we actually get into better and a deeper answer to how does it relate to power work?
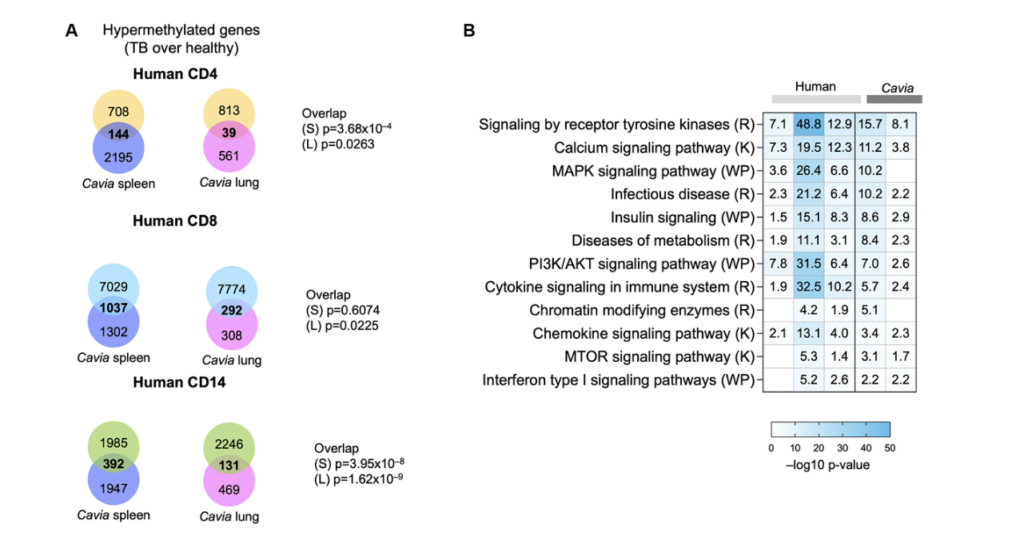
So I had the pleasure of working with Andrew on a paper published in a JCI a few years ago, again, where hyper methylation and persistent hyper methylation persisted, epigenetics carrying was a major theme. And then technically in that paper, we are able to dig through some nifty bioinformatics methods into gene changes in specific cell types. So we looked at CD4 T-cells, CD8, CD14 T-cells in human. And then we look at genes affected by methylation changes in those particular cell types. And then technically we noticed that our two tissues from the guinea pig overlap nicely and significantly with the genes affected by epigenetic changes in the cells. So for example, for the CD4, we have 144 overlaps in spleen and 14 lung. For the CD8 we have almost a thousand in spleen and almost a hundred in lung.
For the CD14 we have 400 and 113, the spleen and lung. So by and large, all of these overlaps are significant. And again, we can see that both the spleen and the lung contribute and teach us something. And then we want again and done a formal enrichment of significant pathways. And we can see that both in the two tissues from the guinea pig and then the pathways from the genes in the three human cell types. We see a lot of the same major players in tuberculosis, for example, immune system, musk kinase, tyrosine kinase, signaling calcium metabolism, and even chromatic modifying enzymes would suggest that there might be further changes in epigenetics that we should study. Andrew I’ll hand this off back to you.
Great. So that brings me to some of my work and for context, a lot of my PhD research was based on developing and applying meta-analysis methods for biological data to study tuberculosis further. And so to do that work, I would often go to the gene expression omnibus, which is hosted by the NCBI and has a wealth of publicly available biological data sets for many diseases, but of course, including tuberculosis. And one of the major things that influences tuberculosis research is that we have these critical TB subpopulations that tend to have many biological differences that can make it difficult to develop consistent diagnostics and therapeutics, et cetera. And two of the major concerns with these subpopulations are those who have HIV comorbidity. And also that TB can present very differently in adults compared to kids. And so for this work specifically, what I was trying to do was identify biological pathways that are similar across these populations, but also that differ across these populations to kind of get a bird’s eye view.
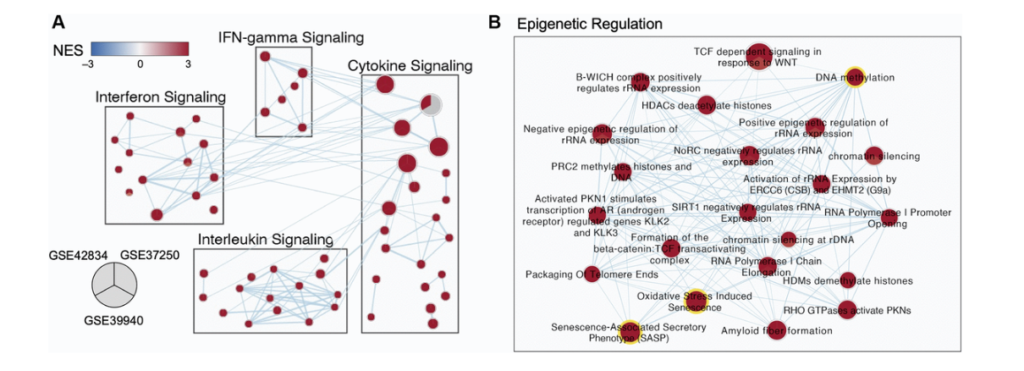
So for this work, we picked three different gene expression, data sets that were collected in whole blood. One of them is in kids. One of them is in adult and does not include any HIV adult patients. And the last is in adults and includes HIV patients. We used Leman to do a differential gene expression analysis and identify our significantly differentiated genes. And then we can rank those gene lists and run them through gene set enrichment analysis, which allows us to identify pathways, which are either enriched or depleted in our population. And then I use the method called enrichment map, which is the next arrow, which allows us to lay these populations over top of each other. To identify at the pathway level, which pathways are consistently enriched or depleted across our different populations. This is an example, a little group of sub pathways showing what that looks like.
And so you can see here each one of these nodes is a pie chart. In each slice of pie corresponds to a different one of those data sets I was just talking about and enriched pathways are shown in red and depleted pathways. Not that you can see any here would be shown in blue. And so you can see here, we are just displaying what I sort of think of as some common frequent flyer pathways for understanding TB infection and biology, including interfering gamma signaling, et cetera. But one of the clusters that really stood out to me and you will see it is we had this whole cluster of epigenetic regulation genes. And this is the work I was presenting at that conference where Andrew and I met and got really excited to talk to each other. Where, and I was finding that pathways and DNA methylation and oxidative distress induced senescence and senescence associated secretary phenotypes were consistently enriched across all three of these TB data sets across these three distinct populations.
And so this sort of orthogonally validated a lot of the research he was already doing and approaching with Cristian. And so we wanted to sort of dive into that at the gene expression level further. So one of the things we did was we designed just a sub z-score for those three different pathways that were just highlighted. So DNA methylation are SAS pathway and our oxidative stress and use senescence pathways. And you can see here that we really have different enrichment of these pathways across either our TB population or asymptomatic control population in all three of these data sets. And it’s quite a distinct pattern that we’re seeing there. And not only that, if we look at the correlation between these scores for these three different pathways, we see that there’s really, really high and strong correlation of these pathways together. Suggesting that they’re somehow working together as part of a TB infection and influencing patients, which is something that continues to be true. Even if we take out some of the overlapping genes that are contained in all of these pathways.
So we had the idea, as I mentioned earlier, that HIV and CMV are known to induce premature epigenetic aging. And when we saw these results, we wanted to look at the overlap between what we saw in humans. So these are the genes that were hyper methylated in humans with TB. And we compared that to a study of age associated, closed chromatin marks that were previously published. And there was a statistical overlap between these, the DNA hypermethylation and the closed chromatin marks and some of the common genes and some of the common pathways are right here. Now we had serum from humans with TB. And we looked to see if common senescence associated proteins were increased. And we looked at approximately six or seven of these proteins, and we found that three of them TNF alpha, CXCL9 and CXCL10 were increased in individuals with TB compared to individuals without TB.
Now, at that point, we applied the Horvath clock, a known DNA methylation clock to the individuals with TB. And we had the individuals with TB when they were originally diagnosed with TB, but we also had a DNA methylation time point for them 12 months from their initial diagnosis. Which is six months after the completion of successful TB therapy. So all of these individuals met the W-H-O criteria for successful TB cure. And you can see that for each case, their DNA methylation age was increased a median of 12.7 years. You can see here, there aren’t a lot of individuals, I believe it’s roughly 12 to 16 individuals per group. And we were lucky in that as we were working on this.
There’s a lot more data, publicly available data, for RNA seek. And so Carly found this and there was this new RNA age clock. So similar to the epigenetic clock, individuals with TB had an RNA age that was approximately 14 years greater than their chronologic age. And this was not significant only for people with TB here in red, but here there was a group of individuals who were asymptomatic and healthy, but in the following year, they went on to develop TB. And you can see that these individuals had an increased cellular age as well.
So previous studies have shown that individuals with increased epigenetic clocks above their chronologic age, where an increased risk of mortality of all cause mortality. One of the future questions we really want to ask since we know that individuals who survive sepsis, pneumonia, TB, and COVID even after propensity matching. They’re at approximately a threefold increased risk of all cause mortality with that increased risk being due to increased heart attacks, strokes, increased risk of cancer and increased risk of respiratory infections. We’d like to know what the relationship between the premature epigenetic aging and the morbidity and mortality. We’re trying to study in vitro. If this is reversible and we have some ideas on ways at which we’re hoping to reverse some of these premature epigenetic aging. And we’re not sure if this is specific to TB. I suspect it’s not, as I stated earlier, there’s some evidence for CMV and HIV that they also in increase cellular aging, but we suspect it’s true for schistosomiasis and bacterial pneumonia. There’s just no data on those right now.
Great. On that last point, I have a quick question about reversibility. And my question is: Certainly this is a great first step in future directions. It’s probably looking at that. Does it reverse on its own though? Do you have any indication of whether this after, I don’t know somebody recovers from TB after a few years, your epigenetic ages or your RNA ages might go back. Do you have any thoughts, expectations, predictions about that?
I have a couple guesses. No, one’s done that. Our study is the longest setting anybody has gone 12 months from initial diagnosis. So we need longer studies. I presented this data to our colleagues in Eswatini 18 months ago, and they immediately said, “All right, well, we need to extend follow up from our current follow up of 12 months all the way to 36 months.” So we’re working on that right now. And we’re trying to get more long term studies here in Houston. We’re enrolling individuals who had pneumonia in the past 36 months so we can go back and look at that. In terms of your other question, in terms of what could reverse premature epigenetic aging. I am excited that certain things like good nutrition might be beneficial for… There’s a large spread… There was a study from Steve Horvaths group that showed that total carotenoids were associated with an epigenetic age lower than your chronologic age.
We have reason to believe that that would be beneficial, more beneficial if you suffer from a serious infection. As Carly showed, there is a strong correlation between oxidative stress and DNA methylation. And we were looking at that specifically because of the previous work that came out of Dr. Horvaths group. And we’re trying to follow that up with in vitro studies. And I would love to follow that up with human studies. How great would it be if we could do something as simple as listen to our grandma’s recommendation to eat a healthy diet? I hope there are some simple drugs that we could use as well that might be able to do it. And all of that just needs to be studied perspectively.
So Brian, as part of what I enjoy by working by informatics is that I get to participate in a lot of intellectual cross pollination. And this means both… Does this method that was developed in this disease, but can be really apply everywhere. And also, well, occasionally I try to learn and pay attention to what my biological collaborators do. So in that sense, on that topic of cross pollination, we’re collaborating and we worked with some folks at the Burke College of Medicine who are essentially using agents like glutathione supplementation, which by essentially, and there’s already a product packaging, glutathione precursors as that type supplements.
And that helps boost the levels of glutathione and how this came to be, how this collaborator is actually working on aging as a side effect in HIV patients. So once treatment improved, where they don’t die early and they’re able to leave productive lives, but then the next phenotype to deal is premature aging. And some of those agents have been shown to work well in elderly people bringing back some cognitive function and everything. So we’re trying to see if we can do projects. And Andrew is very keen to see where this particular agent can it then help with people who has survived to be, but might deal with long term side effects. So again, that’s part of what’s appeals to me to work where I work, because I get to bring things together, see different ideas work from our field to another. So that’s exciting.
Yeah. Well talk about cross pollination. I think I was on that Horvath paper with looking at carotenoids. And then the part of my question was because we’ve looked at longitudinal stability of the different clocks epigenetically across decades of people. And part of the exciting thing about your paper here is that I always think in most of the populations, we look at we’re looking at their average change in their lifestyle over long periods of time. And we might not have the specificity to capture small changes, but also people don’t change their habits all that much. And so why would you expect their epigenetic ages and the aging rates to change? The exciting thing about this paper is this is a strong perturbation to the system as in the form of tuberculosis, and that’s why I’m curious about the persistence of it and whether that lasts and looking at other strong biological factors that… Seeing how that affects the epigenetic agents is really exciting.
So Brian, something that we learned from our working in environmental exposures is that we have seen aging plasticity come into play in the contest of early light BPA exposure and this was reported in Metro communication a few years ago. We are seeing it now in liver after early life [inaudible] exposure. And I think one of the themes that emerge is that aging plasticity is programming all of us. As such, that’s something that will change and is there, and then exposures to environmental, the factors, but infections and others, there is a whole vulnerability right there. So if different interactions can latch on to that and work with that. That’s something that unfortunately is probably pre-programmed as a vulnerability in all of us. I mean, normally just helped us deal with the challenge of… Just chronological challenges. But technically some of these factors, and tuberculosis is one of them, can definitely hijack that and lead to some side effects. And again, we’re seeing it also outside of the disease models that Andrew and Carly have reported here.
So how do you guys see this playing out? Lets kind of assume that tuberculosis and other infectious diseases accelerates aging. Is there a way to prevent that? I guess one possible scenario could be, we find that if you treat it early enough or prevent it, you save a lot of lives, a lot of person years. What are some other scenarios and possible implications that our audience members should think as possibilities in the future?
We are very curious about the metabolic epigenetic communication. We’re calling them the three metabolic, epigenetic trio stats, three mechanisms by which metabolism induced epigenetic changes. And at least in our preliminary in vitro experiments, we are excited to see that if we delay and inhibit that communication, we can delay the scar formation. We need better models to test. If it can actually reverse it as well. That’s one place I’m hoping to see where we can actually manipulate and address these scars. If that doesn’t work, there are direct epigenetic modifying drugs, which currently are still very much global and not targeted the way that we want them to be. And certainly there’s plenty of people working on developing more targeted epigenetic modifiers, but also at the public health level.
This could have pretty significant prognostic implications. So for example, the global mortality for tuberculosis is nine percent. So if you suffer from tuberculosis during therapy, you have a nine percent mortality. There was a meta-analysis of over 40,000 individuals who had survived tuberculosis. And in that meta-analysis 16 percent of them passed away in the following two years, that’s a very large mortality. And again, that mortality was due to cardiovascular disease, cancer, and recurrent respiratory infections, as the top three causes. We need to know if there are certain epigenetic marks that predict who’s going to have one of those three problems, because then we might be able to do something about it, but those studies haven’t yet been done. So we need those longterm studies.
And Brian, one of the other areas were our research can help. So again, in the cross pollination, I think I got my training in cancer bioinformatics and then say breast cancer. Subtypes was a huge revolutionary discovery and that from care. So in that informal paper… So Andrew and I let them just got published a few months ago. A manuscript paper in European Respiratory Journal. Were we essentially identified to tuberculosis endotypes. Show that they have potential clinical implications and they respond differently potentially to commonly use drugs. So on that note, I think perhaps one of the studies we can do and we can see if we can… Can we identify groups of people like endotypes in the tuberculosis infected that might respond differently to aging? So the same way that we see that they respond potentially different drugs, they might have different agent trajectories or technically we can go in a more direct, with the right study and go in a more direct search, where we say, “Look, are there groups of people that are under the same protectively bacteria load and everything.” Still, (does it) lead to responses?
I mean, yeah. Because I think the ideal case scenarios, we see people that are pretty much immune to this aging disruption. And if we can find some people that’s… We want to study to understand what makes them special and learn from it. But no, that would be ideal in a potentially more plausible scenario. If we see that we have people that likely respond to aging, but are much more prone to acceleration, we want to identify them if we can early and perhaps intervene more forcefully or more aggressively. So again, we’re using this endotype concept and we’re hoping that it’ll have implications both in a patient specific treatment in TB, but also maybe teach us something about aging. At this moment, we’re more of the basic research stage, but we’re hoping that it can help address this challenges down the line.
Pure speculations, but that’s good to know what directions we can move towards. So let me end with giving you guys an opportunity to ask for what kind of collaborators, what kind of data sets would you like? What are some things that you would like someone to solve for you so that you can move your research forward? And I’ll like to hear from all three of you. So maybe Andrew, you want to start?
I would love to see large data sets for different types of infections. There’s so few data sets for common viral infections, common bacterial infections, whats the overlap in epigenetic changes between a helmet infection, a mycobacterial infection, a bacterial infection, a viral infection. We don’t know that and better data sets. That would be someone like Carly could tear that and answer that really quickly. I’d love to see, I’d love to see that addressed.
Carly, Cristian.
Yeah. I’d like to echo in any study, making your data available to do additional validation analysis like these. Just has so much benefit to the community, but it’s not just making your data available. It’s also making the metadata available, because I cannot tell you how many times I find a data set that looks like it could be really rich and it could help us answer a question, but I can’t tell what processing steps have happened already.
I’m missing definitions for some of the variables. I’m missing little things. Like one of the struggles we had with this identifying in RNA seek data set to use was that they hadn’t published the known ages of patients. And so we were only able to find the one data set that had done that. And we’re so glad they did because it added so much richness to this, but making that kind of, not just the biology data, but the clinical data and also information about how you’ve cleaned the data available and putting it out there for people. Is really going to move, feels like this forward. And it gives so much extra power to scientists who are trying to work on these interdisciplinary questions and answer.
So Brian, I mean you made the point that, so some of my, what you said speculation and the regard to, but regarding to endotypes and clinical trial is not now because, so on the note, we actually had to collaborate with the group from Germany who had transcriptomics and follow up clinical data, and we are able to demonstrate that our endotypes stratified by time to capture conversion, by cure rate, by death. So we are able to bring some of it and demonstrate a clinical relevance, physician relevance… we have used many, many data sets on the literature and then unfortunately we are always limited. Technically one of the lessons that I think Andrew took to heart and based on his activity, he might be better able to implement in the… He’s involved in planning and executing some large scale tuberculosis.
And the whole plan is to derive both eventually omics of different sorts, but also to compile and make available a risk clinical data. Because then we all focus ultimately, as we discover something, I care about endotypes and we will care about the immune system scaring, but other people I care about other things. I mean, ultimately we teach… The reason they’re all different and the reason isn’t to care about it for this. That’s actually part of the reason that we do so much in science. So to enable everybody to pursue their own hypothesis or interest, talking with Carly, we need to have more access to rich and informative metadata and yeah.
Infectious disease field is, as Carly stated, we are far behind the oncology field. So more than a decade ago, the NIH really invested in the TCGA the cancer genome Atlas. And there’s not a single cancer that hasn’t been improved based on that Atlas, we have nothing like that for infectious diseases. That Atlas has clinical outcomes, rich, rich epidemiology, rich clinical information, linked to every omic that you would want to study. And there’s nothing like that for infectious diseases. So whether it’s breast cancer, prostate cancer, leukemia, glioblastoma. Mining that data has identified new therapies and I’m confident that infectious diseases would similarly find the common differences targeted therapy, precision medicine in the same way that cancer has benefited from the TCGA.
And well, just to accelerate the conversation on that last point. So there are groups out there that can run epigenetics very easily, but then access to tissues is the challenge or access to the metadata? Is that latter part solved yet? Are there people, or is there a consortium where there is a gathering of tissue and information?
Not that I know of.
No, not that I’m aware of either.
Well that seems like a great first step, whoever succeeds Francis Collins. Well with that, thank you all for your time. I know there were multiple time zones that we were just coordinating around, but really exciting. And thank you for sharing your thoughts and your paper. Congratulations on the publication and best of luck with all the rest of your research.
Click here to read the full paper published by Aging (Aging-US).
Longevity & Aging Series Episode 3: Dr. Steve Horvath – Epigenetic Clocks
Longevity & Aging Series Episode 2: Dr. Steve Horvath’s Special Collection in Aging
Longevity & Aging Series Episode 1: Drs. Alex Zhavoronkov and Frank Pun
AGING (AGING-US) VIDEOS: YouTube | LabTube | Aging-US.com
—
Aging (Aging-US) is an open-access journal that publishes research papers bi-monthly in all fields of aging research and other topics. These papers are available to read at no cost to readers on Aging-us.com. Open-access journals offer information that has the potential to benefit our societies from the inside out and may be shared with friends, neighbors, colleagues, and other researchers, far and wide.
For media inquiries, please contact [email protected].